Variability in Angelman Syndrome
What Does This Mean, and Why Does This Matter?
An important question that may arise with the diagnosis of Angelman Syndrome, and that may persist is “what will my child be like?”. This is a very natural question following a diagnosis as families and caregivers seek to understand the implications of the diagnosis.
This article will address some basic scientific concepts upon which variability in genetic syndromes is grounded, as well as discuss the effects that the ideas and realities of genetic variability actually have for both the child and the family living with Angelman Syndrome (often referred to as “AS”).
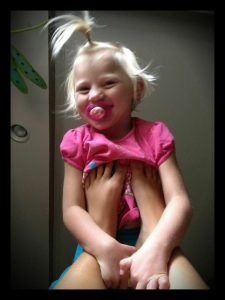 What Will My Child Be Like?
What Will My Child Be Like?
You may have noticed that your child is not achieving expected milestones and sought help with a medical professional. Due to the fact that it is a rare disorder, it is common for doctors to not at first suspect or test for Angelman Syndrome. As such,
a diagnosis is sometimes not forthcoming. However, some diagnoses of Angelman Syndrome are made before one year of age.
When a parent is told the diagnosis is Angelman syndrome and they are made familiar with the characteristics of this disorder, such as seizures, sleep problems, non-verbal communication, severe cognitive impairment and more, it is quite natural to wonder what the future may hold.
Early diagnosis can be beneficial. First, one has more time to come to terms with what it means to raise a child with special needs. This can be intimidating for there are many factors to consider including the following: special education; doctors and specialists; daily living; and long term living options. An early diagnosis also means an early start of therapies and access to services, which has been shown to be of enormous benefit to the child. A late diagnosis is equally important. While the parents/caregivers may already suspect that their child has special needs, getting a genetic diagnosis of AS opens the door to a wide base of knowledge and strategies that are effective in AS, as well as providing access to a community of others with shared experiences.
What will your child be like? It is a natural question to wonder if they will be able to sit up or walk. You may ask, “Will my child talk or use sign language? How will they play with other children? Will my child go to school? Will he/she be able to ride a bike? How will my child’s development progress and how does that affect plans for the future?”
While no one can predict milestone attainment, we do know that not all children with AS experience the same degree and severity of symptoms. For example, while most children with AS will experience some form of seizure activity, some children with AS never have seizures.

In order to understand the manner in which these differences occur, we need to explore additional aspects affecting the genetics of Angelman Syndrome. (Please visit the Genetics page on the FAST website for an in-depth review of the basic genetics of AS). Two key concepts that can help explain variation in Angelman Syndrome are genetic penetrance and genetic expression.
Genetic Penetrance
Penetrance is a term used in genetics to explain the likelihood that a gene (genotype) will express an associated trait or appearance (phenotype). Populations expressing a genetic variation (like the loss of UBE3A function seen in Angelman Syndrome) are described by associating that genotype with certain characteristics. In Angelman Syndrome, the characteristics define the syndrome. For example, 70% of deletion children will have a sleep disorder, or 1% of all AS individuals will have mosaicism.

Penetrance can be described as:
- Complete penetrance - all individuals having the gene variation have clinical symptoms associated with the variation.
- Highly penetrant- the trait caused by the gene variation is almost always apparent.
- Reduced penetrance - some individuals may not express the trait, although they carry the gene variation.
- Low penetrance - a trait will rarely be apparent even when the gene variation exists. In this case it is often difficult to differentiate between genetic and environmental factors as a ‘cause’ of a trait.
The current body of research suggests that Angelman Syndrome falls into the realm of complete penetrance. That is, anyone with a mutation that affects maternal
UBE3A expression will present with characteristics of Angelman Syndrome. However, there are different
expressions of the phenotype among individuals. If we take into account the other 99.9% of inherited genes, as well as differentiated environmental influences, it is quite normal to find differences in abilities and development among those diagnosed with Angelman Syndrome.
Genetic Expression
Angelman Syndrome is just that....a syndrome. If developmental milestones and abilities are plotted, a normal bell shaped curve of distribution occurs. Some individuals will have minimal self-help abilities and need maximum care, while others may have more developed self-help skills and need less assistance.
While there is a recognized relationship between genotype and the
severity of phenotype in Angelman Syndrome, the correlation is not definite. For example, there may be two children with Angelman Syndrome that share the same class of deletion size, yet there are manifest differences in milestone attainment and expression of abilities.
The following concepts can help explain this:
- In Angelman Syndrome, there is a functional loss of the maternal UBE3A gene. When the maternal copy of the gene is nonfunctional, (or non expressive), Angelman Syndrome results. Without that functional copy of the gene, a myriad of problems occur, which among others results in a brain that cannot process experiences correctly, or lay down memories in typical fashion. This translates into cognitive delay, non verbal behavior, seizures, and more. The paternal UBE3A gene is still present, but remains ‘silent’, which is the normal state of the paternal gene.
- Generally, children that are born with a deletion of the UBE3A region (encompassing more genes than just the UBE3A gene), tend to have greater challenges in terms of motor development and seizure control. The reason that deletion positive children are generally more severely affected is likely due to the loss of expression of many other genes in
the region. Other genotypes of AS (UPD, ICD, UBE3A mutation) show a tendency to be more mildly impacted than deletion positive types, again likely due to the fact that people with these genotypes have not lost additional genes in the Angelman region of chromosome 15.
- Environmental influences also play a large part in determining the developmental progress of children born with AS. Getting timely medical care and follow-up, experiencing physical, occupational and speech therapy and having a firm plan for schooling that supports the particular needs of the child can account for some variations in development, all other factors aside.
The message here is that your child is both the product of their ~29,000 other normally functioning genes, as well as their life experiences. A child who is diagnosed with Angelman Syndrome due to a genetic deletion may also look very different in terms of characteristics than those diagnosed with a non- deletion error (such as uniparental disomy), or an
UBE3A mutation, or some type of mosaicism.
However, the genotype is not alone in determining the phenotype. While intense therapies such as those provided by Early Intervention Programs are normally directed toward children from birth to three, obtaining resources and providing therapies shown to be effective in Angelman Syndrome are important even if a child is diagnosed later in life. Effective therapies, and positive environmental influences, have been shown to increase developmental abilities, as well as enhance the quality of life for many individuals with AS. Medical professionals and local human services agencies can provide guidance toward resources that result in meaningful developmental enhancement.

While there is no present ‘cure’ for the genetic defect in Angelman Syndrome, it has been shown that physical, occupational, and speech therapies are beneficial for children with developmental delay, regardless of genotype or present developmental attainment.
How Genetic Variation in Angelman Syndrome affects Your Child
As discussed above, there are variations in the presentation of Angelman Syndrome from individual to individual due to the combination of genetics and environment. As a parent or caregiver of an individual with AS, it is important to remember that nothing is set in stone. It is a mistake to presume that all of the characteristics associated with this syndrome will present in each individual to the degree described by clinicians for the syndrome in general.

In assessing developmental achievement or progress in such areas as “when did the child start to sit up, walk, potty train, play games, etc...”, the variance is great, even among AS children of similar genotype. There are a few key factors to keep in mind that impact developmental attainment as well as “phenotype”, or appearance:
Genetic Penetrance and Expression: While Angelman Syndrome may fall into the realm of complete penetrance, the expression of the other genes in the genome play strong roles in your child’s abilities and personality.
Epigenetic Regulation: Outside influences (environment, diet, medications, etc...) may influence the level of skills a person will have through enhancing inherent potentials and managing developmentally disabling medical conditions with medical intervention.
Seizure Disorder: This can have a significant impact on the development of an individual and should be closely monitored by a licensed medical professional. Medications and dietary modifications have been shown to alleviate epilepsy and should be discussed with the child’s licensed medical professional.
Therapy and Services: There is ample scientific evidence that points to the success of Early Intervention in terms of physical, occupational, and speech therapy for persons with disabilities. Therapies for older individuals can also be effective in enhancing skills and the quality of life.
Diet and Nutrition: Like any individual, a person with Angelman Syndrome needs a nutritious diet. Given the feeding and reflux issues many individuals with AS deal with, it is beneficial to seek advice from dieticians and GI specialists If a child is unable to take foods orally, parents
should consult with a medical professional about a G-tube, which delivers nutrients through a surgically manufactured opening in the stomach.
Establish a ‘circle of support’ for the individual with Angelman Syndrome as well as for the parents/caregivers. This can include caregivers, friends, caseworkers, teachers, medical professionals, therapists and family members. The idea is to build a support network for the individual and parents/caregivers, so that as the individual grows, there are multiple resources to draw upon when needed. For example, it could mean something as simple as needing good respite care while the primary caregiver does household errands, or something as complex as arranging for special care when traveling. Also, having people around the individual that care about his/her welfare and growth, will go a long way toward allowing the individual to achieve his/her fullest potential through early childhood, the teenage years, and into adulthood.

A final thought to impart, is that while a person may be born with a loss of function of the one gene,
UBE3A (the “Angelman” gene), she or he is,
however, born with thousands of other fully functional genes. This, more than anything else, will determine “what your child will look like”. Yes, an Angelman Syndrome diagnosis may be life-changing, but many other factors will influence development and determine the overall quality of life experience. Foremost are the variety of enriched environment experiences that allow for learning, growth and achievement.
The best thing any parent can do, is to get informed. Find a great doctor. Seek out other parents and join an Angelman Syndrome organization that best meets your values. Become an advocate and aim high. Small miracles happen every day. It makes good sense, to leave the door open to experience them.
(All of the individuals pictured in this article have Angelman Syndrome.)
All articles, quotes and materials are copyrighted ©. Copyright protected worldwide all rights reserved. This article can be copied or distributed for educational purposes only, but must not be altered in form or content.
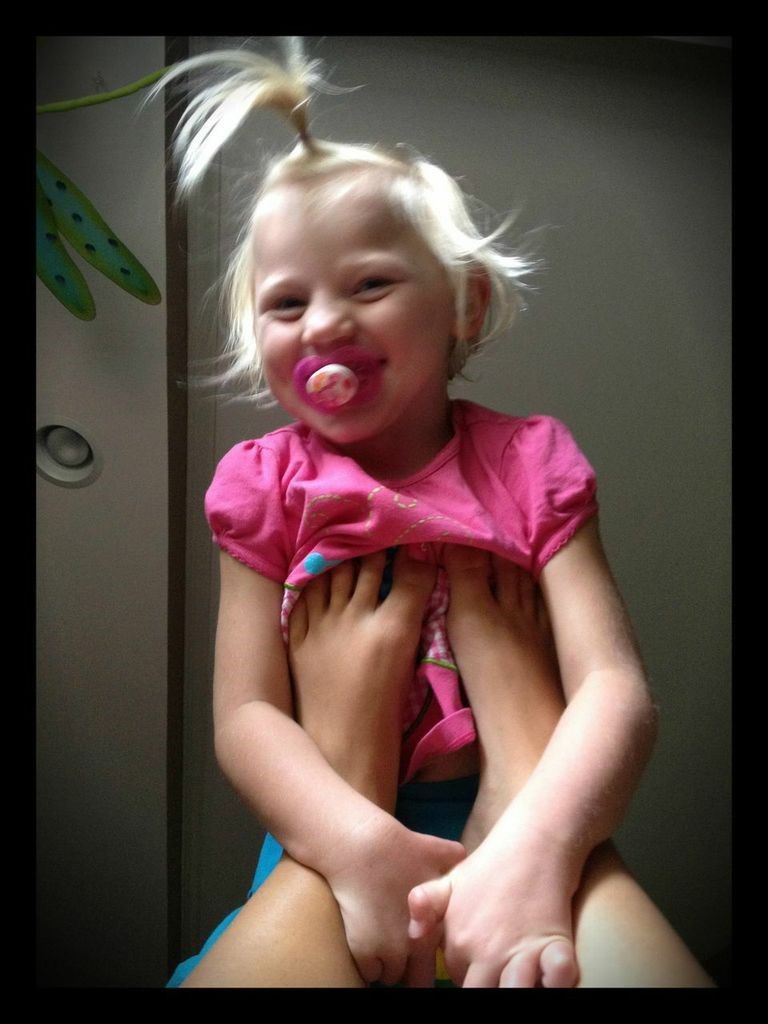 What Will My Child Be Like?
You may have noticed that your child is not achieving expected milestones and sought help with a medical professional. Due to the fact that it is a rare disorder, it is common for doctors to not at first suspect or test for Angelman Syndrome. As such,
a diagnosis is sometimes not forthcoming. However, some diagnoses of Angelman Syndrome are made before one year of age.
When a parent is told the diagnosis is Angelman syndrome and they are made familiar with the characteristics of this disorder, such as seizures, sleep problems, non-verbal communication, severe cognitive impairment and more, it is quite natural to wonder what the future may hold.
Early diagnosis can be beneficial. First, one has more time to come to terms with what it means to raise a child with special needs. This can be intimidating for there are many factors to consider including the following: special education; doctors and specialists; daily living; and long term living options. An early diagnosis also means an early start of therapies and access to services, which has been shown to be of enormous benefit to the child. A late diagnosis is equally important. While the parents/caregivers may already suspect that their child has special needs, getting a genetic diagnosis of AS opens the door to a wide base of knowledge and strategies that are effective in AS, as well as providing access to a community of others with shared experiences.
What will your child be like? It is a natural question to wonder if they will be able to sit up or walk. You may ask, “Will my child talk or use sign language? How will they play with other children? Will my child go to school? Will he/she be able to ride a bike? How will my child’s development progress and how does that affect plans for the future?”
While no one can predict milestone attainment, we do know that not all children with AS experience the same degree and severity of symptoms. For example, while most children with AS will experience some form of seizure activity, some children with AS never have seizures.
What Will My Child Be Like?
You may have noticed that your child is not achieving expected milestones and sought help with a medical professional. Due to the fact that it is a rare disorder, it is common for doctors to not at first suspect or test for Angelman Syndrome. As such,
a diagnosis is sometimes not forthcoming. However, some diagnoses of Angelman Syndrome are made before one year of age.
When a parent is told the diagnosis is Angelman syndrome and they are made familiar with the characteristics of this disorder, such as seizures, sleep problems, non-verbal communication, severe cognitive impairment and more, it is quite natural to wonder what the future may hold.
Early diagnosis can be beneficial. First, one has more time to come to terms with what it means to raise a child with special needs. This can be intimidating for there are many factors to consider including the following: special education; doctors and specialists; daily living; and long term living options. An early diagnosis also means an early start of therapies and access to services, which has been shown to be of enormous benefit to the child. A late diagnosis is equally important. While the parents/caregivers may already suspect that their child has special needs, getting a genetic diagnosis of AS opens the door to a wide base of knowledge and strategies that are effective in AS, as well as providing access to a community of others with shared experiences.
What will your child be like? It is a natural question to wonder if they will be able to sit up or walk. You may ask, “Will my child talk or use sign language? How will they play with other children? Will my child go to school? Will he/she be able to ride a bike? How will my child’s development progress and how does that affect plans for the future?”
While no one can predict milestone attainment, we do know that not all children with AS experience the same degree and severity of symptoms. For example, while most children with AS will experience some form of seizure activity, some children with AS never have seizures.
 In order to understand the manner in which these differences occur, we need to explore additional aspects affecting the genetics of Angelman Syndrome. (Please visit the Genetics page on the FAST website for an in-depth review of the basic genetics of AS). Two key concepts that can help explain variation in Angelman Syndrome are genetic penetrance and genetic expression.
Genetic Penetrance
Penetrance is a term used in genetics to explain the likelihood that a gene (genotype) will express an associated trait or appearance (phenotype). Populations expressing a genetic variation (like the loss of UBE3A function seen in Angelman Syndrome) are described by associating that genotype with certain characteristics. In Angelman Syndrome, the characteristics define the syndrome. For example, 70% of deletion children will have a sleep disorder, or 1% of all AS individuals will have mosaicism.
In order to understand the manner in which these differences occur, we need to explore additional aspects affecting the genetics of Angelman Syndrome. (Please visit the Genetics page on the FAST website for an in-depth review of the basic genetics of AS). Two key concepts that can help explain variation in Angelman Syndrome are genetic penetrance and genetic expression.
Genetic Penetrance
Penetrance is a term used in genetics to explain the likelihood that a gene (genotype) will express an associated trait or appearance (phenotype). Populations expressing a genetic variation (like the loss of UBE3A function seen in Angelman Syndrome) are described by associating that genotype with certain characteristics. In Angelman Syndrome, the characteristics define the syndrome. For example, 70% of deletion children will have a sleep disorder, or 1% of all AS individuals will have mosaicism.
 Penetrance can be described as:
Penetrance can be described as:
 While there is no present ‘cure’ for the genetic defect in Angelman Syndrome, it has been shown that physical, occupational, and speech therapies are beneficial for children with developmental delay, regardless of genotype or present developmental attainment.
How Genetic Variation in Angelman Syndrome affects Your Child
As discussed above, there are variations in the presentation of Angelman Syndrome from individual to individual due to the combination of genetics and environment. As a parent or caregiver of an individual with AS, it is important to remember that nothing is set in stone. It is a mistake to presume that all of the characteristics associated with this syndrome will present in each individual to the degree described by clinicians for the syndrome in general.
While there is no present ‘cure’ for the genetic defect in Angelman Syndrome, it has been shown that physical, occupational, and speech therapies are beneficial for children with developmental delay, regardless of genotype or present developmental attainment.
How Genetic Variation in Angelman Syndrome affects Your Child
As discussed above, there are variations in the presentation of Angelman Syndrome from individual to individual due to the combination of genetics and environment. As a parent or caregiver of an individual with AS, it is important to remember that nothing is set in stone. It is a mistake to presume that all of the characteristics associated with this syndrome will present in each individual to the degree described by clinicians for the syndrome in general.
 In assessing developmental achievement or progress in such areas as “when did the child start to sit up, walk, potty train, play games, etc...”, the variance is great, even among AS children of similar genotype. There are a few key factors to keep in mind that impact developmental attainment as well as “phenotype”, or appearance:
Genetic Penetrance and Expression: While Angelman Syndrome may fall into the realm of complete penetrance, the expression of the other genes in the genome play strong roles in your child’s abilities and personality.
Epigenetic Regulation: Outside influences (environment, diet, medications, etc...) may influence the level of skills a person will have through enhancing inherent potentials and managing developmentally disabling medical conditions with medical intervention.
Seizure Disorder: This can have a significant impact on the development of an individual and should be closely monitored by a licensed medical professional. Medications and dietary modifications have been shown to alleviate epilepsy and should be discussed with the child’s licensed medical professional.
Therapy and Services: There is ample scientific evidence that points to the success of Early Intervention in terms of physical, occupational, and speech therapy for persons with disabilities. Therapies for older individuals can also be effective in enhancing skills and the quality of life.
Diet and Nutrition: Like any individual, a person with Angelman Syndrome needs a nutritious diet. Given the feeding and reflux issues many individuals with AS deal with, it is beneficial to seek advice from dieticians and GI specialists If a child is unable to take foods orally, parents
should consult with a medical professional about a G-tube, which delivers nutrients through a surgically manufactured opening in the stomach.
Establish a ‘circle of support’ for the individual with Angelman Syndrome as well as for the parents/caregivers. This can include caregivers, friends, caseworkers, teachers, medical professionals, therapists and family members. The idea is to build a support network for the individual and parents/caregivers, so that as the individual grows, there are multiple resources to draw upon when needed. For example, it could mean something as simple as needing good respite care while the primary caregiver does household errands, or something as complex as arranging for special care when traveling. Also, having people around the individual that care about his/her welfare and growth, will go a long way toward allowing the individual to achieve his/her fullest potential through early childhood, the teenage years, and into adulthood.
In assessing developmental achievement or progress in such areas as “when did the child start to sit up, walk, potty train, play games, etc...”, the variance is great, even among AS children of similar genotype. There are a few key factors to keep in mind that impact developmental attainment as well as “phenotype”, or appearance:
Genetic Penetrance and Expression: While Angelman Syndrome may fall into the realm of complete penetrance, the expression of the other genes in the genome play strong roles in your child’s abilities and personality.
Epigenetic Regulation: Outside influences (environment, diet, medications, etc...) may influence the level of skills a person will have through enhancing inherent potentials and managing developmentally disabling medical conditions with medical intervention.
Seizure Disorder: This can have a significant impact on the development of an individual and should be closely monitored by a licensed medical professional. Medications and dietary modifications have been shown to alleviate epilepsy and should be discussed with the child’s licensed medical professional.
Therapy and Services: There is ample scientific evidence that points to the success of Early Intervention in terms of physical, occupational, and speech therapy for persons with disabilities. Therapies for older individuals can also be effective in enhancing skills and the quality of life.
Diet and Nutrition: Like any individual, a person with Angelman Syndrome needs a nutritious diet. Given the feeding and reflux issues many individuals with AS deal with, it is beneficial to seek advice from dieticians and GI specialists If a child is unable to take foods orally, parents
should consult with a medical professional about a G-tube, which delivers nutrients through a surgically manufactured opening in the stomach.
Establish a ‘circle of support’ for the individual with Angelman Syndrome as well as for the parents/caregivers. This can include caregivers, friends, caseworkers, teachers, medical professionals, therapists and family members. The idea is to build a support network for the individual and parents/caregivers, so that as the individual grows, there are multiple resources to draw upon when needed. For example, it could mean something as simple as needing good respite care while the primary caregiver does household errands, or something as complex as arranging for special care when traveling. Also, having people around the individual that care about his/her welfare and growth, will go a long way toward allowing the individual to achieve his/her fullest potential through early childhood, the teenage years, and into adulthood.
 A final thought to impart, is that while a person may be born with a loss of function of the one gene, UBE3A (the “Angelman” gene), she or he is,
however, born with thousands of other fully functional genes. This, more than anything else, will determine “what your child will look like”. Yes, an Angelman Syndrome diagnosis may be life-changing, but many other factors will influence development and determine the overall quality of life experience. Foremost are the variety of enriched environment experiences that allow for learning, growth and achievement.
The best thing any parent can do, is to get informed. Find a great doctor. Seek out other parents and join an Angelman Syndrome organization that best meets your values. Become an advocate and aim high. Small miracles happen every day. It makes good sense, to leave the door open to experience them.
(All of the individuals pictured in this article have Angelman Syndrome.)
A final thought to impart, is that while a person may be born with a loss of function of the one gene, UBE3A (the “Angelman” gene), she or he is,
however, born with thousands of other fully functional genes. This, more than anything else, will determine “what your child will look like”. Yes, an Angelman Syndrome diagnosis may be life-changing, but many other factors will influence development and determine the overall quality of life experience. Foremost are the variety of enriched environment experiences that allow for learning, growth and achievement.
The best thing any parent can do, is to get informed. Find a great doctor. Seek out other parents and join an Angelman Syndrome organization that best meets your values. Become an advocate and aim high. Small miracles happen every day. It makes good sense, to leave the door open to experience them.
(All of the individuals pictured in this article have Angelman Syndrome.)