Genomic Imprinting Does Not Reduce the Dosage of UBE3A in Neurons
Summary by Allyson Berent, DVM, DACVIM
Angelman syndrome (AS) is caused by loss of the maternally inherited copy of the UBE3A gene. Like most genes, the UBE3A gene is present on both alleles of chromosome 15, the one inherited from the mother (maternal allele) and the one inherited from the father (paternal allele); however, UBE3A is different from roughly 99.9% of the genes in the genome in one very particular important way - the paternal UBE3A allele is turned-off in the brain, whereas the maternal UBE3A allele is on. But, in all the other organs of the body, both the maternal and paternal alleles of UBE3A are on. This explains why AS is caused by mutations/deletions affecting the maternally inherited allele of UBE3A but not the paternally inherited allele of UBE3A. In the brain, where UBE3A plays perhaps its most important role, the maternal UBE3A allele is typically on and the paternal UBE3A allele is typically off. In all other organs of the body, both parental UBE3A alleles are on. This is a normal process and occurs in all people and almost all animal species.
 Why the paternal UBE3A allele is specifically turned-off in the brain is unknown. This phenomenon where one allele is on and the other allele is off is called genomic imprinting, and it happens at only a few genes in the genome. Nevertheless, it is believed to be very important, as almost all imprinted genes are associated with a human disease. One theory is that genomic imprinting evolved to reduce the amount of protein made by the gene, which is referred to as the dosage model of genomic imprinting. Quite simply, the idea is that one allele will make about half as much protein as two alleles. The notion that imprinting is used to lower the amount of UBE3A protein made in the brain makes sense, especially since it’s known that too much UBE3A is associated autism spectrum disorder.
As part of their effort to understand how and why the UBE3A gene is imprinted in the brain, the Dindot laboratory tested the hypothesis that imprinting functions to lower, or regulate, the amount of UBE3A made in the brain. This has been assumed but never tested.
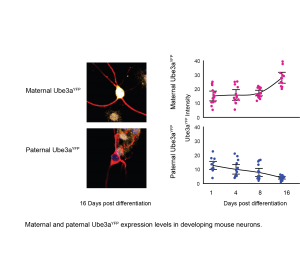
Thus, scientists in the Dindot laboratory used various techniques to measure the levels of UBE3A in almost all organs of the human and mouse body. They also measured UBE3A levels in individual neurons in the mouse brain as well as in individual neurons outside the brain, for example in the colon. They also went as far as to measure the amount of UBE3A protein made from the maternal and paternal UBE3A alleles while the father’s copy of UBE3A is actually being turned off in neurons!
Their findings were surprising. In all cases, the expression levels of UBE3A in normal individuals were either higher or the same in the brain than those in other organs, even though the brain had half the amount of the UBE3A being read. More importantly, they found that while the paternal UBE3A allele is being turned off during the development of neurons in the brain, the amount of UBE3A protein made by the maternal allele compensates for loss of the paternal UBE3A allele, maintaining a constant level of UBE3A protein in the developing neurons. Collectively, their results showed that in the brain the maternal UBE3A allele somehow produces more UBE3A protein to make-up for the loss created by the paternal UBE3A allele being turned off.
So why is this important? Well, the answer is somewhat complicated. The FIRE team, as well as several other groups, have been trying to find ways to turn on the paternal UBE3A allele as a therapy for AS. This approach to wake-up a silent gene is indeed possible, and it has been done for UBE3A using 3 or 4 different methods. But, this approach has never been used for a gene in a clinical setting, so it’s important to understand why UBE3A is imprinted in the brain in order to know what might happen if it is turned-on in an individual with AS. Altogether, this study by Hillman et al., is one of a series of FAST-funded research projects to help safely develop new therapies to turn-on the paternal UBE3A allele as a therapy for AS.
Angelman syndrome is cause by the functional loss of the maternal copy of the gene UBE3A. This gene is normally present on both the maternal (mother’s) and the paternal (father’s) allele of chromosome 15. When the copy on the maternal side is missing, or nonfunctional, the condition is known as AS.
In the central nervous system (CNS=brain and spinal cord) the maternal copy is typically functional and the gene is able to be read, or transcribed. The protein of UBE3A is then produced, or translated. This protein is necessary for many neurologic functions. The paternal copy is read and “translated” into a protein throughout the body, but in the CNS it is “imprinted” or “silenced” and not read. Therefore, the brain requires the copy from the maternal allele to function properly.
The specific aim of this study was to evaluate if loss of imprinting of the paternal allele (turn on the paternal copy of UBE3A) in the brain would increase the dosage of UBE3A, in an attempt to prove if the imprinting of the paternal UBE3A gene evolved as a dose-regulating mechanism. The UBE3A transcript and protein levels were evaluated in both brain and peripheral tissues of the body (thymus, heart, liver, lung, etc…) in both mice and humans.
The results of this study found no correlation between the imprinting status and the dosage of UBE3A in any cells of the body, in both mice and humans. In addition, during embryogenesis it was found that the maternal Ube3a protein levels actually increased when the paternal Ube3A levels decreased, fully compensating for the loss of expression of the paternal Ube3a gene in neurons.
Based on these results it is speculated that the imprinting of the UBE3A gene in humans on the paternal allele does not function to reduce the dosage of UBE3A in neurons but rather to help regulate some other, yet still unknown, aspect of gene expression or protein function. In addition, the peripheral levels of UBE3A in other organs where the paternal gene is not imprinted are likely not severely impacted by a missing copy of the gene on one of the alleles containing UBE3A.
We might be able to extrapolate from this information the notion that activation of the paternal gene of UBE3A in humans may not lead to overactivation of UBE3A as seen in other conditions like Dup15q further supporting these novel therapeutics for Angelman syndrome.
To access the full text publication, click here.
Why the paternal UBE3A allele is specifically turned-off in the brain is unknown. This phenomenon where one allele is on and the other allele is off is called genomic imprinting, and it happens at only a few genes in the genome. Nevertheless, it is believed to be very important, as almost all imprinted genes are associated with a human disease. One theory is that genomic imprinting evolved to reduce the amount of protein made by the gene, which is referred to as the dosage model of genomic imprinting. Quite simply, the idea is that one allele will make about half as much protein as two alleles. The notion that imprinting is used to lower the amount of UBE3A protein made in the brain makes sense, especially since it’s known that too much UBE3A is associated autism spectrum disorder.
As part of their effort to understand how and why the UBE3A gene is imprinted in the brain, the Dindot laboratory tested the hypothesis that imprinting functions to lower, or regulate, the amount of UBE3A made in the brain. This has been assumed but never tested.
Thus, scientists in the Dindot laboratory used various techniques to measure the levels of UBE3A in almost all organs of the human and mouse body. They also measured UBE3A levels in individual neurons in the mouse brain as well as in individual neurons outside the brain, for example in the colon. They also went as far as to measure the amount of UBE3A protein made from the maternal and paternal UBE3A alleles while the father’s copy of UBE3A is actually being turned off in neurons!
Their findings were surprising. In all cases, the expression levels of UBE3A in normal individuals were either higher or the same in the brain than those in other organs, even though the brain had half the amount of the UBE3A being read. More importantly, they found that while the paternal UBE3A allele is being turned off during the development of neurons in the brain, the amount of UBE3A protein made by the maternal allele compensates for loss of the paternal UBE3A allele, maintaining a constant level of UBE3A protein in the developing neurons. Collectively, their results showed that in the brain the maternal UBE3A allele somehow produces more UBE3A protein to make-up for the loss created by the paternal UBE3A allele being turned off.
So why is this important? Well, the answer is somewhat complicated. The FIRE team, as well as several other groups, have been trying to find ways to turn on the paternal UBE3A allele as a therapy for AS. This approach to wake-up a silent gene is indeed possible, and it has been done for UBE3A using 3 or 4 different methods. But, this approach has never been used for a gene in a clinical setting, so it’s important to understand why UBE3A is imprinted in the brain in order to know what might happen if it is turned-on in an individual with AS. Altogether, this study by Hillman et al., is one of a series of FAST-funded research projects to help safely develop new therapies to turn-on the paternal UBE3A allele as a therapy for AS.
Angelman syndrome is cause by the functional loss of the maternal copy of the gene UBE3A. This gene is normally present on both the maternal (mother’s) and the paternal (father’s) allele of chromosome 15. When the copy on the maternal side is missing, or nonfunctional, the condition is known as AS.
In the central nervous system (CNS=brain and spinal cord) the maternal copy is typically functional and the gene is able to be read, or transcribed. The protein of UBE3A is then produced, or translated. This protein is necessary for many neurologic functions. The paternal copy is read and “translated” into a protein throughout the body, but in the CNS it is “imprinted” or “silenced” and not read. Therefore, the brain requires the copy from the maternal allele to function properly.
The specific aim of this study was to evaluate if loss of imprinting of the paternal allele (turn on the paternal copy of UBE3A) in the brain would increase the dosage of UBE3A, in an attempt to prove if the imprinting of the paternal UBE3A gene evolved as a dose-regulating mechanism. The UBE3A transcript and protein levels were evaluated in both brain and peripheral tissues of the body (thymus, heart, liver, lung, etc…) in both mice and humans.
The results of this study found no correlation between the imprinting status and the dosage of UBE3A in any cells of the body, in both mice and humans. In addition, during embryogenesis it was found that the maternal Ube3a protein levels actually increased when the paternal Ube3A levels decreased, fully compensating for the loss of expression of the paternal Ube3a gene in neurons.
Based on these results it is speculated that the imprinting of the UBE3A gene in humans on the paternal allele does not function to reduce the dosage of UBE3A in neurons but rather to help regulate some other, yet still unknown, aspect of gene expression or protein function. In addition, the peripheral levels of UBE3A in other organs where the paternal gene is not imprinted are likely not severely impacted by a missing copy of the gene on one of the alleles containing UBE3A.
We might be able to extrapolate from this information the notion that activation of the paternal gene of UBE3A in humans may not lead to overactivation of UBE3A as seen in other conditions like Dup15q further supporting these novel therapeutics for Angelman syndrome.
To access the full text publication, click here.
Why the paternal UBE3A allele is specifically turned-off in the brain is unknown. This phenomenon where one allele is on and the other allele is off is called genomic imprinting, and it happens at only a few genes in the genome. Nevertheless, it is believed to be very important, as almost all imprinted genes are associated with a human disease. One theory is that genomic imprinting evolved to reduce the amount of protein made by the gene, which is referred to as the dosage model of genomic imprinting. Quite simply, the idea is that one allele will make about half as much protein as two alleles. The notion that imprinting is used to lower the amount of UBE3A protein made in the brain makes sense, especially since it’s known that too much UBE3A is associated autism spectrum disorder.
As part of their effort to understand how and why the UBE3A gene is imprinted in the brain, the Dindot laboratory tested the hypothesis that imprinting functions to lower, or regulate, the amount of UBE3A made in the brain. This has been assumed but never tested.
Thus, scientists in the Dindot laboratory used various techniques to measure the levels of UBE3A in almost all organs of the human and mouse body. They also measured UBE3A levels in individual neurons in the mouse brain as well as in individual neurons outside the brain, for example in the colon. They also went as far as to measure the amount of UBE3A protein made from the maternal and paternal UBE3A alleles while the father’s copy of UBE3A is actually being turned off in neurons!
Their findings were surprising. In all cases, the expression levels of UBE3A in normal individuals were either higher or the same in the brain than those in other organs, even though the brain had half the amount of the UBE3A being read. More importantly, they found that while the paternal UBE3A allele is being turned off during the development of neurons in the brain, the amount of UBE3A protein made by the maternal allele compensates for loss of the paternal UBE3A allele, maintaining a constant level of UBE3A protein in the developing neurons. Collectively, their results showed that in the brain the maternal UBE3A allele somehow produces more UBE3A protein to make-up for the loss created by the paternal UBE3A allele being turned off.
So why is this important? Well, the answer is somewhat complicated. The FIRE team, as well as several other groups, have been trying to find ways to turn on the paternal UBE3A allele as a therapy for AS. This approach to wake-up a silent gene is indeed possible, and it has been done for UBE3A using 3 or 4 different methods. But, this approach has never been used for a gene in a clinical setting, so it’s important to understand why UBE3A is imprinted in the brain in order to know what might happen if it is turned-on in an individual with AS. Altogether, this study by Hillman et al., is one of a series of FAST-funded research projects to help safely develop new therapies to turn-on the paternal UBE3A allele as a therapy for AS.
Angelman syndrome is cause by the functional loss of the maternal copy of the gene UBE3A. This gene is normally present on both the maternal (mother’s) and the paternal (father’s) allele of chromosome 15. When the copy on the maternal side is missing, or nonfunctional, the condition is known as AS.
In the central nervous system (CNS=brain and spinal cord) the maternal copy is typically functional and the gene is able to be read, or transcribed. The protein of UBE3A is then produced, or translated. This protein is necessary for many neurologic functions. The paternal copy is read and “translated” into a protein throughout the body, but in the CNS it is “imprinted” or “silenced” and not read. Therefore, the brain requires the copy from the maternal allele to function properly.
The specific aim of this study was to evaluate if loss of imprinting of the paternal allele (turn on the paternal copy of UBE3A) in the brain would increase the dosage of UBE3A, in an attempt to prove if the imprinting of the paternal UBE3A gene evolved as a dose-regulating mechanism. The UBE3A transcript and protein levels were evaluated in both brain and peripheral tissues of the body (thymus, heart, liver, lung, etc…) in both mice and humans.
The results of this study found no correlation between the imprinting status and the dosage of UBE3A in any cells of the body, in both mice and humans. In addition, during embryogenesis it was found that the maternal Ube3a protein levels actually increased when the paternal Ube3A levels decreased, fully compensating for the loss of expression of the paternal Ube3a gene in neurons.
Based on these results it is speculated that the imprinting of the UBE3A gene in humans on the paternal allele does not function to reduce the dosage of UBE3A in neurons but rather to help regulate some other, yet still unknown, aspect of gene expression or protein function. In addition, the peripheral levels of UBE3A in other organs where the paternal gene is not imprinted are likely not severely impacted by a missing copy of the gene on one of the alleles containing UBE3A.
We might be able to extrapolate from this information the notion that activation of the paternal gene of UBE3A in humans may not lead to overactivation of UBE3A as seen in other conditions like Dup15q further supporting these novel therapeutics for Angelman syndrome.
To access the full text publication, click here.